Introduction
Water Molecular Dynamics Simulation is a physics-driven C++ application that recreates the emergence of water molecules from individual oxygen and hydrogen atoms. By computing bonded and non-bonded forces at each time step, the program delivers a scientifically grounded yet visually engaging demo of molecular behavior in an interactive 3D environment.

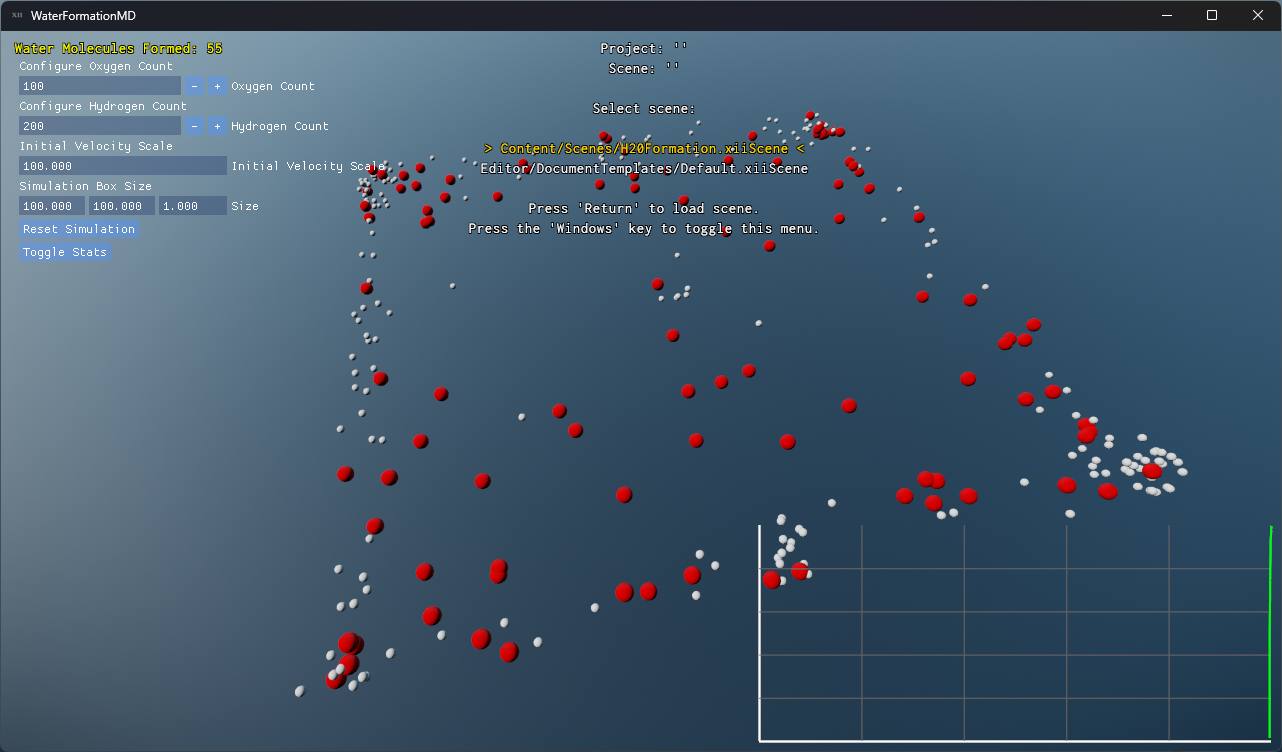
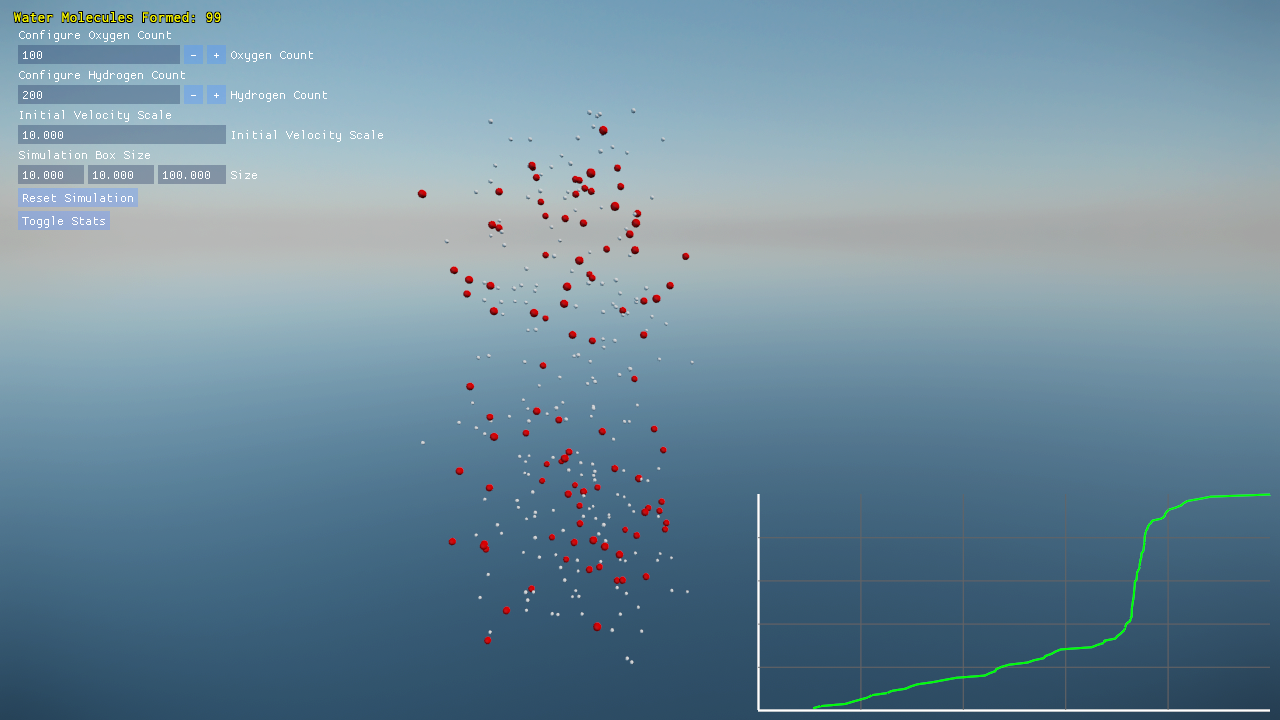
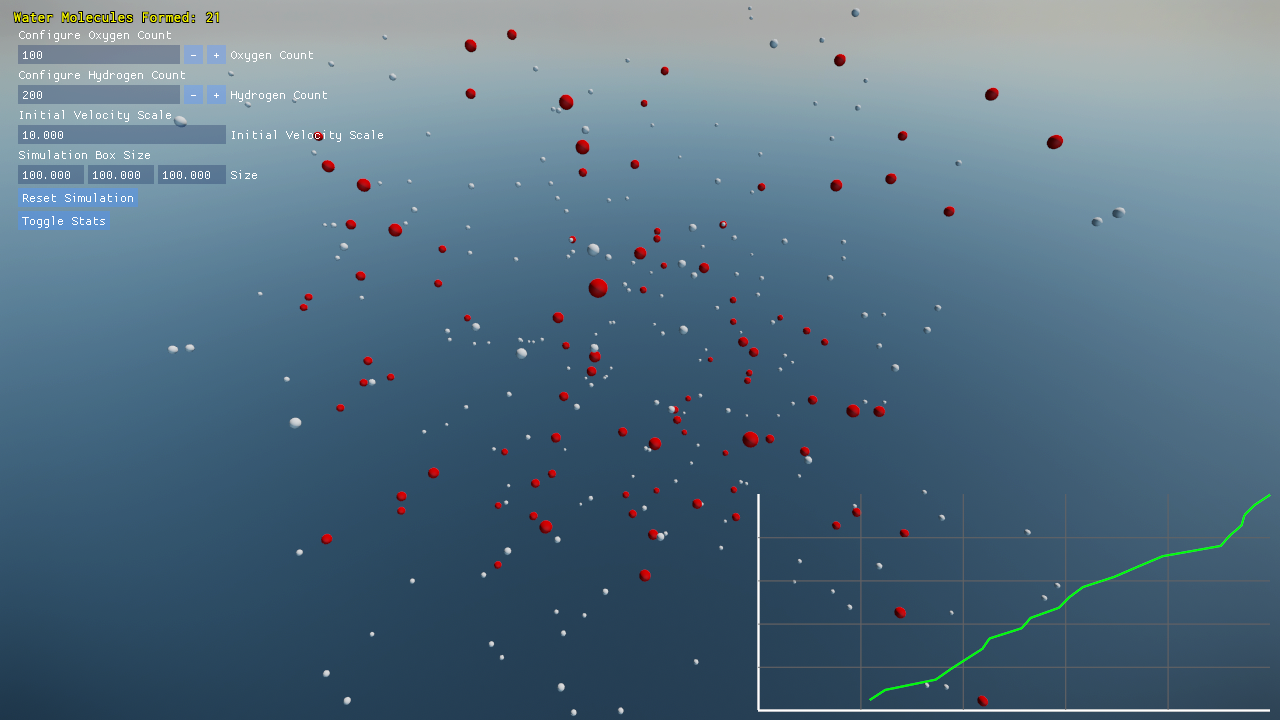
Project Overview
- Purpose: Offer an educational, real-time window into the forces shaping water at the molecular scale.
- Engine Core: Custom C++ simulation engine with ImGui for live parameter tweaking.
- Key Algorithms:
- Harmonic bond stretching for O–H bonds
- Harmonic angle bending for H–O–H geometry
- Lennard-Jones and Coulomb potentials for non-bonded interactions
- Platforms: Windows and Linux.
Features
- Dynamic molecular interactions governed by physics-based potentials.
- Real-time ImGui interface to adjust:
- Oxygen and hydrogen atom counts
- Initial velocity scale
- Simulation box dimensions
- Performance overlay with FPS, compute timings, and debug console.
- Built-in plotting of water molecule formation progress over time.
Settings Panel
| Parameter | Default | Description |
|---|---|---|
| Oxygen atoms | 100 | Number of O atoms spawned at simulation start |
| Hydrogen atoms | 200 | Number of H atoms spawned at simulation start |
| Initial velocity scale | 1.0 | Scales random initial velocities of all atoms |
| Simulation box size | 10×10×10 | World bounds for atom positions and reflections |
Use the sliders and numeric inputs in the ImGui panel to experiment with density and kinetic energy on the fly.
Visualization and Debugging
A debug overlay provides real-time statistics:
- Frame rate display (toggle with F5).
- Kernel timing breakdowns.
- On-screen graphs plotting the count of complete H₂O molecules over time.
Screenshots and performance data help you understand both the molecular dynamics and the engine’s efficiency.
Controls
- Camera: WASD + mouse to fly around the simulation box.
- UI Toggle Keys:
- F2: Open/close debug console (Tab for auto-complete).
- F5: Show/hide FPS counter.
- F12: Capture a high-resolution screenshot.
These shortcuts let you navigate, inspect, and document the simulation without interrupting the main loop.
How It Works
At each integration step, forces on every atom are summed from:
Bonded Forces
- Harmonic bond stretching: Keeps O–H bonds near their equilibrium length.
- Harmonic angle bending: Enforces the natural H–O–H angle.
Non-Bonded Forces
- Lennard-Jones potential for van der Waals interactions.
- Coulomb’s law for electrostatic attraction/repulsion.
Positions and velocities update via simple velocity-Verlet integration, ensuring stability at small time steps.
Future Work
- Refine angle-bending model for more precise H₂O geometry.
- Add explicit thermostat to control temperature.
- Support multiple species (ions, contaminants).
- Expand visualization modes (surface mesh, volumetric density).
License
This project is released under the MIT License. See LICENSE for details.